

The Dark Matter Project
The completion of the first fully sequenced human genome in 2003 came with a surprise: protein coding genes comprise only 2 percent of total DNA, equating to approximately 20,000 genes, considerably fewer than expected. The key to our multicellular complexity presumably resides in the remaining 98 percent of non-coding DNA, or “dark matter,” the function of which remains largely elusive. Current research reveals that individual variation in the non-coding genome plays key roles in many diseases such as neurodegenerative diseases, heart disease, and cancer, to name a few. While Genome-Wide Association Studies (GWAS) are a first step in discovering variants correlated with disease predisposition, there are limitations to this low-resolution methodology making pinpointing exact disease variant susceptibility traits difficult Which combinations of variants contribute to disease manifestation? Which variants are protective against disease? How does the structure and context of regulatory elements affect the expression of target genes? Interpreting the role of dark matter is a challenging undertaking, but rapid advancements in DNA synthesis and assembly technologies, which vastly improve speed, fidelity, and cost, allows us to start probing the answers to these questions.
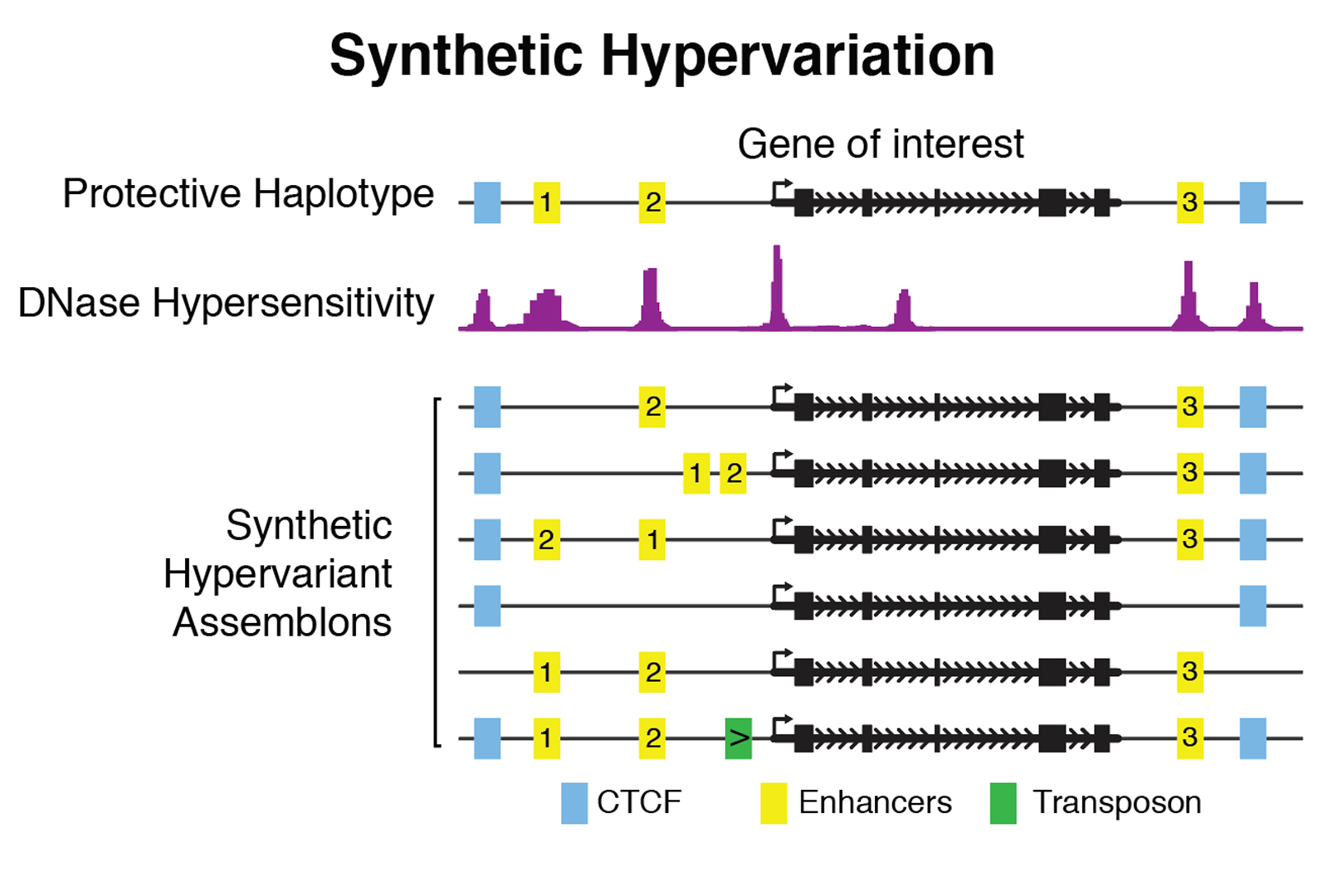
We are supported in part by a NIH/NHGRI-funded Center for Excellence in Genome Sciences (CEGS). We aim to decipher the function of substantial portions of the non-coding genome. We are taking a bottom-up approach to design and write defined variation in these sequences, in a manner that allows us to answer questions relating to variation in specific functional elements to phenotypic outcomes. We have three distinct objectives, namely design and assembly of thousands of large (greater than 100kbp) DNA constructs we call “assemblons,” their faithful and site-specific delivery to the genomes of relevant cell types, and finally, functional readouts of the variants. We are focusing mostly on mouse and human embryonic stem cells as experimental platforms. We delineate two unique applications of our technology: with synthetic hypervariation, we can manipulate individual functional elements (for example, enhancers, CTCF sites) within our assemblies to interrogate their functions when removed, relocated or otherwise modified.
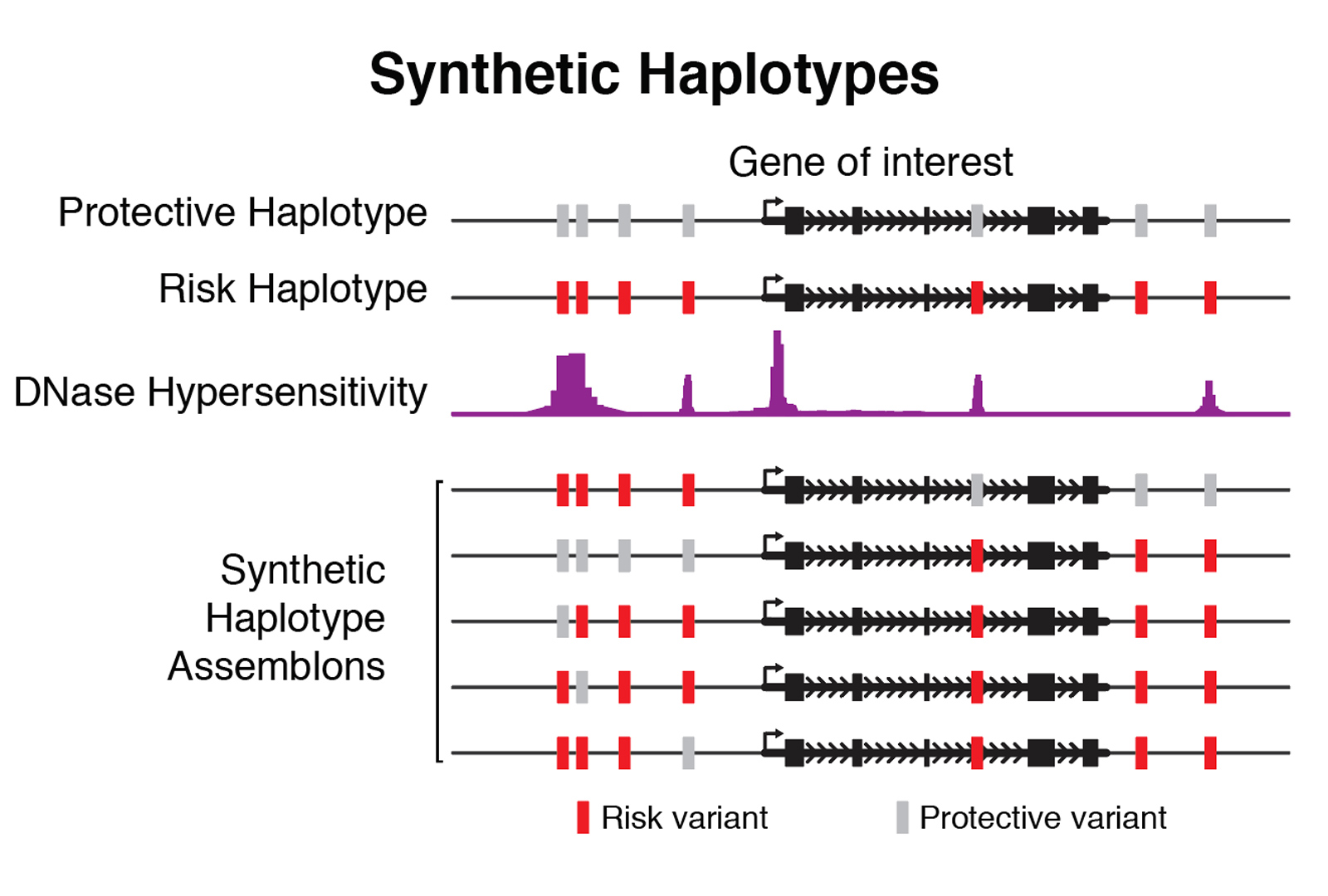
The second primary application is in the assembly of synthetic haplotypes. We mix and match naturally occurring genetic variants (for example, SNPs), generating haplotypes with variant combinations absent from natural populations, facilitating identification of functional variants or combinations thereof that lead to phenotypic outcomes. Our systematic assembly, delivery, and phenotyping scheme will further allow us to develop generalized modeling approaches to describe the functions of various non-coding elements in mammalian genomes, enabling new strategies for design and assembly of functional synthetic DNA.
Current loci under investigation include HTRA1, HoxA, IRF5, α-globin, Sox2, ANK1, and Xist. For more information and to propose your favorite locus, visit the Dark Matter Project.
